バゾプレシンと体液量調節
1.はじめに
バゾプレシン(AVP)は下垂体後葉から分泌されるアミノ酸9個からなるペプチドホルモンである。主として視床下部の視索上核(SON)、室傍核(PVN)の大細胞性ニューロンで産生され、軸策輸送で下垂体後葉へ運ばれた後、血中へ分泌され、ホルモンとして作用する。室傍核には小細胞性ニューロンもあり、ここで産生されたAVPは下垂体前葉をはじめ、脳内の様々な部位に投射され、神経伝達物質などとして作用している。AVPの受容体(R)は細胞膜7回貫通のG蛋白共役型受容体であり、3種類の受容体が知られている。V1aRは血管平滑筋、肝臓、血小板に分布し、それぞれ血管収縮、グリコーゲン分解、血小板凝集作用を示す。またV1aRは脳内にも広く分布しており、記憶、情動に関与するとされている。V1bRは下垂体前葉のACTH産生細胞に存在し、PVNの小細胞性ニューロンで合成されたAVPが下垂体門脈に分泌されることによりV1bRを介して下垂体前葉からのACTH分泌を促進する。V2Rは腎尿細管に存在し、AVPはV2Rを介して水チャネルであるアクアポリン2(AQP2)の発現と管腔側細胞膜への細胞内輸送を増強させることにより尿細管での水再吸収を増加させて、抗利尿作用を現わす。また、血管内皮細胞にも存在し、第Ⅷ因子やvon Willebrand因子の分泌を促進して血液凝固機構に関与する。
2.AVPの分泌調節
AVPの分泌は主として血漿浸透圧及び循環血液量の変化による調節を受けている。浸透圧受容器は第三脳室前腹側壁の終盤脈絡器官(OVLT)や脳弓下器官(SFO)などに存在する。血漿浸透圧の上昇がこれらの部位で感知され、視床下部PVN、SONのAVPニューロンにその情報が伝達されて、AVPの分泌が促進される。視床下部のAVPニューロン自身にも浸透圧感受性があるとされるが生理的意義は明らかではない。浸透圧変化に対するAVPの分泌変化は非常に鋭敏であり、血漿浸透圧の1%の上昇でもAVPの分泌が増加する。また、浸透圧によるAVP分泌には閾値があり、通常血漿浸透圧 280 mOsm/kgH2O程度以下ではAVPの分泌はshut offされている。血漿浸透圧がこの閾値以上になると、AVP分泌が直線的に増加する。また、血漿浸透圧 が290 mOsm/kgH2O程度以上になると、口渇中枢が刺激されて飲水行動が惹起され、AVPの分泌増加とともに体液量保持に寄与する。浸透圧刺激によるAVP分泌の感度は体液量の程度にはあまり依存しないが、脱水状態では閾値がやや左方移動すると考えられている1。一方、浸透圧上昇と体液量減少は相乗的にAVP分泌を刺激するという成績もある2。なお、実際にはAVPの分泌刺激となるのは、浸透圧(osmolality)ではなく張度(tonicity)であり、浸透圧受容器では細胞内外の張度差による細胞容積の変化を何らかの機構で感知していると想定されている3。尿素は浸透圧の構成要素ではあるが、細胞膜を比較的自由に透過し、細胞内外の張度差の形成には関与しないため、有効なAVP分泌刺激とはならない。ブドウ糖も、インスリン存在下では、細胞膜透過性が高いため、有効なAVP分泌刺激にはならないと考えられている4。
血液量の減少及び血圧の低下もAVPの分泌刺激となる。血液量の増加は左房壁の伸展受容器などの容積受容器で感知され上行性の迷走神経によって延髄弧束核に伝えられる。一方、血圧の上昇は頚動脈洞や大動脈弓にある圧受容器で感知され、それぞれ舌咽、迷走神経により延髄弧束核に伝えられる。これらの経路は通常状態ではAVP分泌を持続的に抑制しており、血液量減少あるいは血圧低下時にこれらの抑制が解除されることでAVPの分泌が亢進する。弧束核からは延髄腹外側部A1領域、青班核A6領域などを介してのノルアドレナリン作動性神経が視床下部のPVN、SONに投射されAVP分泌に対し促進的に作用する。血液量あるいは血圧変化に対するAVPの分泌変化は浸透圧の場合ほど鋭敏ではなく、AVP分泌を惹起するには10~15%の血液量減少あるいは血圧低下が必要とされるが、それを超える変化に対しては指数関数的なAVP分泌の増加がみられる。
嘔気も強力なAVPの分泌刺激である5。嘔吐中枢である延髄の最後野(area postrema)を介すると想定される。嘔気に嘔吐を伴うことは必要としないが、逆に嘔気を伴わない嘔吐もAVPの分泌刺激になりうる。他の生理的刺激として、頚動脈小体や大動脈小体には化学受容器があり、低酸素血症を感知するとそれぞれ舌咽あるいは迷走神経を介して脳幹のノルアドレナリン作動性ニューロンを活性化することによりAVP分泌を刺激する。また、低血糖もAVPの分泌刺激となるがその機序はよくわかっていない。
3.AVPによる体液量調節
現在の哺乳類の細胞内環境は生物が誕生した当時の原始の海に近いともいわれている。その後、岩塩から溶け出した塩分により海水の塩分濃度は上昇し、現在の海水の塩分濃度は約3%と哺乳類の細胞外液の約3倍となっている。生物がまだ海水中で生活していたときは、この高い海水の塩分濃度によって脱水状態とならないための機構が必要であった。サメなどの軟骨魚類では体内に尿素を蓄えることで血漿浸透圧を高め、海水の高浸透圧に対抗している。一方、硬骨魚類は海水中の塩分を効果的に排泄し体内の塩分濃度を下げる方向に進化した。そのためには塩分を効率よく排泄する仕組みが必要であり、AVP(硬骨魚類ではバソトシン)もこれに関与していたと考えられている。しかし、魚類の腎臓にはAQP2がなく、V2Rの存在も最近になってわかったことであり、尿細管での水の再吸収にはAVPは関与していなかったと想定される6。その後生物が陸に上がるようになると、今度はいかに脱水にならないように体内に水を保持するかという機構が重要となった。このため、AVPは役目を変え、抗利尿ホルモンとして腎臓での水の再吸収を促進するのが主な生理的作用となったと推察される。
AVPの腎尿細管での主な作用はAQP2の調節による水の再吸収増加であるが、実際には後述するように、Naや尿素の再吸収も促進する。これは腎髄質高浸透圧の維持に重要である。すなわち、AVPはAQP2の発現を促進して尿細管細胞膜の水の通りをよくするとともに、尿細管管腔内と腎間質との浸透圧格差を形成することで、水の移動すなわち再吸収を促進している。
また、体内ではNaの代謝と水の代謝は密接な関係があり、Naの再吸収が増えて体内Na量が増えれば体内水分量が増加する。このNa調節にもっとも重要なホルモンはアルドステロンであるが、AVPは傍糸球体装置のmacula densaのV1a受容体を介してレニンの分泌を促進し、レニン-アンジオテンシンーアルドステロン(RAA)系を活性化する。すなわち、AVPはアルドステロン分泌の亢進を介したNa再吸収の促進によっても体液量の増加に関与すると考えられる。
このようにAVPは単にAQP2の発現を増やすだけではなく、多方面の作用により水の再吸収を促進し、体液量の維持に貢献している(図1)。以下に、その詳細を述べる。
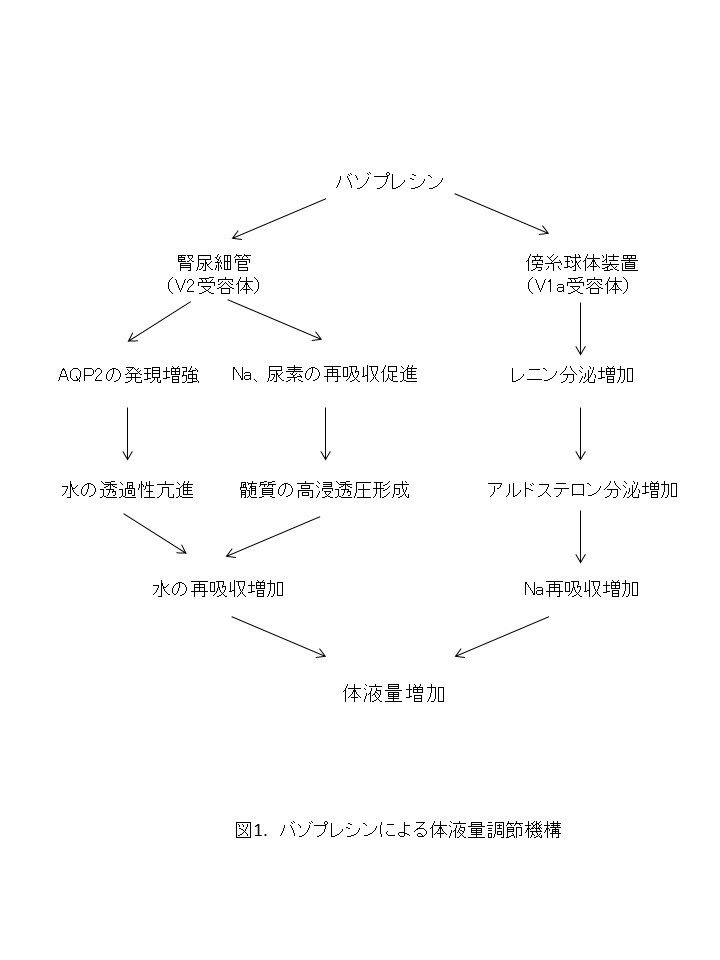
1) 水の再吸収
AQPは水チャネルのファミリーであり、6回膜貫通型のチャネルで四量体を形成している。人ではAQP0からAQP12の13種類が知られている。このうち、AVPによって調節され、AVPの抗利尿作用に直接関与するのはAQP2である。細胞膜に存在するAQP2は常に開いていて、浸透圧勾配に応じて水を透過すると考えられている。AVPによるAQP2の調節には発現量の調節と、管腔側膜へのtraffickingという細胞内輸送調節の2つがある。前者は効果発現に数時間を有するのでlong-term調節と言われ、後者は30分以内に効果発現がみられるのでshort-term調節と言われる。尿細管管腔側膜のAQP2を介して細胞内に入った水は基底膜側のAQP3、AQP4を介して血液中に取り込まれる。
a)AQP2の発現量調節(long-term調節)
AVPは腎集合管主細胞の基底膜側に存在するV2Rに結合し、Gsを活性化する。その結果、adenylyl cyclaseが活性化されて細胞内のcAMP濃度が上昇する。AQP2遺伝子のpromoter領域にはcAMP responsive element(CRE)が存在するため、CREBやAP-1の活性化を介してAQP2 mRNAの転写を促進し、その結果、AQP2 mRNA量が増加し、AQP2蛋白が増加する7。なお、AVPによるシグナルはAQP2 mRNAの安定化には関与しないという報告がある。また、細胞内cAMP濃度の上昇はAQP2蛋白の分解も促進すると言われるが、全体としては産生増加によりAQP2蛋白の増加につながると考えられている8。
b)AQP2の細胞内輸送調節(short-term調節)
上述のようにAVPは腎尿細管においてV2Rを介して細胞内cAMP濃度を上昇させる。それにより、protein kinase A(PKA)が活性化された結果、AQP2のC末端のserineのリン酸化が修飾されることで、細胞骨格アクチンフィラメントの脱重合が触媒され、AQP2の尿細管管腔側膜への集積が増加する。なかでも、serine 256 (S256)のリン酸化が最も重要と考えられており、S256が常にリン酸化された状態にしておくと、AVPの刺激なしでもAQP2は尿細管管腔側膜に留まる9。一方、管腔側膜から細胞内へのrecyclingにはこのS256の脱リン酸化は必要ないと考えられている。それ以外にもS264、S269のリン酸化とS261の脱リン酸化がAQP2の管腔側膜への輸送を促進すると言われている10。また、S256のリン酸化はAQP2の水透過性自体も亢進させるという報告がある11。
2)Naの再吸収
AVPは腎において、V2Rを介したNa再吸収作用と、V1aRを介したNa排泄作用の両方の作用を有する。通常状態でAVPが高濃度でないときは以下のようにV2Rを介して2種類のNaチャネルを活性化することによるNa再吸収作用が前面に出る。一方、AVPは腎血管平滑筋のV1a受容体を介して腎の血行動態や糸球体濾過率(GFR)に影響し、Naの再吸収を阻害してNa排泄を促進する作用も有する。このため、脱水時などAVP濃度が高いときにはAVPの作用はNa排泄に傾くこともある12。
a)上皮性Naチャネル(ENaC)
AVPは遠位尿細管から集合管においてV2Rに結合し、ENaCを活性化してNaの再吸収を促進する。ENaCはα,β,γの3量体からなるNaチャネルで遠位尿細管から集合管に分布する。Amirolideで阻害を受けamirolide-sensitve Na channelともいわれる。アルドステロンの標的チャネルでもあり、アルドステロンのNa再吸収作用は主にこのチャネルを介して行われる。一方、Na利尿ペプチドのNa排泄作用の一部はこのENaCの阻害によるとされる。AVPによるENaCの活性化は、主として管腔側膜へのtraffickingの効果によるものと考えられているが、それ以外にAVPはENaCの各subunitの発現量を増加させることも報告されている13。AVPによるENaCのtrafficking調節の機序の詳細は明らかではないが、ENaCの抑制因子であるNedd4-2(neural precursor cell expressed developmentally down-regulated 4-2)をPKAがリン酸化して阻害する機序が想定されている12。なお、アルドステロンはmineralocorticoid receptorに結合してSgk (serum and glucocorticoid-inducible kinase)を活性化し、その結果同様にNedd4-2を阻害してENaCを活性化することでNa再吸収を促進すると考えられている。
b) Na-K-2Cl 共輸送体(NKCC2)
V2RはHenleのループの太い上行脚(TAL)にも存在することが知られている。AVPはこのV2Rを介してNKCC2を活性化し、TALでのNaの再吸収を促進する。NKCC2はBumetanideやfurosemideなどのループ利尿剤で阻害されることからbumetanide-sensitive co-transporter-1 (BSC-1)とも呼ばれる。NKCC2はNKCC1と高い相同性を有するが、NKCC1はヒトでは結腸などで発現がみられる14。NKCC2の遺伝子異常による不活性変異はBartter症候群を生じることが知られている。AVPによるNKCC2の活性調節の詳細は明らかではないが、管腔側へのtraffickingとリン酸化による活性化が重要と考えられている15。また、AVPはNKCC2の発現量を増加させることも報告されている16。しかし、ヒトの実験で、amirolide投与によりAVPのV2R agonistであるDDAVPによるNa再吸収作用がほぼ完全に抑制されたことから、少なくともヒトにおいては、AVPによる尿細管でのNa再吸収作用はENaCを介するものが主体であって、NKCC2の役割は限定的という報告もある17。
3) 尿素の再吸収
AVPはV2Rを介し、遠位尿細管から集合管のUTA-1などの尿素輸送体を活性化して尿素の再吸収を促進する。その機序はリン酸化による管腔側膜への移動によるものと考えられる。AVPのこの作用にはPKAによるUTA-1のserine 486と499のリン酸化が重要と報告されている。また、Epac (exchange protein activated by cAMP)の活性化の関与も示唆されている18。なお、脱水時にはBUN/Cr比が増大することが知られている。脱水によるGFRの低下は尿素とクレアチニンをともに上昇させるが、クレアチニンは尿細管で再吸収されないのに対し、尿素は脱水時に再吸収が亢進するためであり、この再吸収増加にはAVPの関与も示唆される。
4) RAA系の活性化
V1aRは広く血管平滑筋に分布し血圧調節には重要であることが以前から知られていたが、その体液量調節における意義は不明であった。近年、V1a knock-out マウスを用いた解析で、V1a knock-out マウスでは野生型マウスに比べ体液量が減少していることが示された19。その機序を検討してみると、レニン、アルドステロン濃度が減少していた。従来、腎臓の傍糸球体装置では、macula densa細胞におけるnNOSとCOX-2によるNOおよびPGE2産生が顆粒細胞からのレニン分泌を促進することが知られていた。最近、V1aRとnNOS、COX-2の共存が明らかとなり、AVPは腎臓の傍糸球体装置においてmacula densaのV1aRに結合し、nNOSとCOX-2の発現増加を介してレニン分泌を促進し、RAA系を活性化してアルドステロン分泌を増加させることが示唆された。一方、V1aR knock-out マウスでは、傍糸球体装置からのレニン分泌が低下しアルドステロン分泌が減少することが、体液量減少につながると推察された20。また、AVPは副腎皮質のV1aRを介して直接アルドステロンの分泌を刺激することも知られており、V1aR knock-out マウスではこのAVPによる直接的アルドステロン分泌作用が低下していることも報告されている21。
4.AVP分泌異常症と体液量
1)AVP分泌過剰症(SIADH)
正常であれば、飲水や輸液により体内水分量が増加して血漿浸透圧が低下すると、AVPの分泌が抑制され、腎尿細管での水の再吸収が低下して水利尿が増加する。その結果、体内水分量が減少し、血漿浸透圧は回復する。SIADHは、血漿浸透圧が低下しても、何らかの原因によりAVPの分泌が十分抑制されず、水利尿不全から体内水貯留が進行し、希釈性の低Na血症を呈する病態である。しかし、教科書的にSIADHでは浮腫が見られないとなっているように、慢性期では体液量の増加は軽度で約10%以内の増加と言われる。その機序としてAVPの抗利尿作用からのエスケープ現象の存在が知られている。エスケープ現象とは、SIADHでは慢性的なAVPの過剰分泌とそれによる体液貯留により、V2受容体及びAQP2のdown-regulationを生じ、AVPの抗利尿作用が減弱するものである22。V2Rのdown-regulationは高濃度のAVP自体によるものと、体液量増大によるものの両方の機序による。エスケープ現象におけるAQP2のdown-regulationはlong-term調節、すなわち発現量の低下が主体であり、short-term 調節である細胞内traffickingは影響されないという報告がある。AQP2のdown-regulationの機序の詳細はいまだに明らかではないが、我々はSIADHのラットモデルを用いた検討で、SIADHでは体液量増大により腎動脈還流圧が上昇し、その結果腎尿細管でのPGE2やNOの産生が亢進するためであると報告した23。一方、培養腎細胞を用いた検討で、浸透圧刺激がAQP2の発現を増加させることから、低Na血症に伴う腎間質の浸透圧低下もAQP2の発現低下を惹起すると考えられている24。このようなエスケープ現象の存在は、AVPの過剰分泌によって体液量貯留が進行し、低Na血症がさらに悪化することを防ぐための生体の防御機構として機能すると考えられる。
また、我々はAVPの慢性的過剰分泌が生体に及ぼす影響を明らかにするためにAVP過剰発現transgenic rat(TG)を作製し、その水代謝異常を検討した。このTGでは血漿AVP基礎値は13.8 pg/mlと著明高値であったが、尿量の減少は軽度であり、低Na血症は認めなかった25。液体食を負荷後にも血清Na値の低下は軽度であった。腎臓のV2Rの発現が低下していたが、V2R拮抗剤であるOPC31260投与により回復した。さらにOPC31260で前処置をしたのちに液体食を投与すると、血清Naの著明な低下を認めた。すなわち、慢性的なAVPの過剰分泌によりV2Rがdown-regulationされていて、その結果高AVP血症による過剰な抗利尿作用が代償されていると考えられた26。また、基礎状態での血圧は正常ラットと差はなかったが、AVP皮下注に対する昇圧反応や、脱血による血圧低下からの回復が有意に低下していた。V1aR拮抗剤であるOPC21268前処置によりこれらの反応は改善した。このことから、慢性的高AVP血症下では、AVPによる昇圧作用がV1aRのdown-regulationにより代償されていることが推察された27。
2)尿崩症
尿崩症にはAVPの分泌不足による中枢性尿崩症と、AVPの腎作用の減弱による腎性尿崩症がある。いずれもAVPの抗利尿作用の低下により多尿をきたす。典型的な尿崩症では1日の尿量が10~15Lに達することもある。そのままでは高張性脱水状態が引き起こされるが、口渇中枢が正常に機能していれば、体液量減少による血漿浸透圧上昇に応じて飲水量が増大し、それにより尿量の増加は代償されるため、明らかな脱水や高Na血症にはならない。しかし、腫瘍浸潤などで口渇中枢とAVP産生ニューロンが同時に傷害されると、飲水量による代償ができなくなるため、高度の高張性脱水を呈することになる。なお、、まれに腫瘍や炎症などで、視床下部のAVPニューロンは傷害されないが、口渇中枢のみ傷害されることがある。このような場合、浸透圧刺激によるAVP分泌は低下しているが、体液量減少に対するAVP分泌反応は保たれている。このため、高Na血症は呈するが、脱水の程度は軽度で、いわゆる本態性高Na血症の病態となる。最近、視床下部の浸透圧受容器におけるNa濃度センサーと想定されるNaXチャネルに対する自己抗体により、本態性高Na血症を呈した症例が報告された28。
5.おわりに
AVPは腎尿細管において主としてAQP2の発現を調節して水の再吸収を促進するが、AQP2は水チャネルであってポンプのように能動的なものではなく、水の移動の原動力は尿細管腔と腎髄質との浸透圧格差である。AVPは尿細管における尿素やNaの再吸収を促進することで、髄質の高浸透圧の維持にも関与している。またRAA系の活性化とそれによるアルドステロン分泌にも関与する。このようにAVPは多方面の作用により腎尿細管で水とNaの再吸収を促進し、体液量の維持に貢献している。
文献
1. Sharif-Naeini R, Ciura S, Zhang Z, Bourque CW. Contribution of TRPV channels to
osmosensory transduction, thirst, and vasopressin release. Kidney Int 2008; 73:811.
2. Robertson GL, Athar S. The interaction of blood osmolality and blood volume in regulating plasma vasopressin in man. J Clin Endocrinol Metab 1976; 42:613.
3. Stricker EM, Verbalis JG. Interaction of osmotic and volume stimuli in regulation of neurohypophyseal secretion in rats. Am J Physiol 1986; 250:R267.
4. Vokes TP, Aycinena PR, Robertson GL. Effect of insulin on osmoregulation of vasopressin. Am J Physiol 1987; 252:E538.
5. Rowe JW, Shelton RL, Helderman JH, et al. Influence of the emetic reflex on vasopressin release in man. Kidney Int 1979; 16(6):729.
6. 今野紀文. バソトシンV2型受容体の機能からみた硬骨魚類の環境適応. 比較内分泌学2011; 37(141), 89.
7. Hasler U, Leroy V, Martin PY, Féraille E. Aquaporin-2 abundance in the renal collecting duct: new insights from cultured cell models. Am J Physiol Renal Physiol 2009; 297(1):F10.
8. Hasler U, Nielsen S, Féraille E, Martin PY. Posttranscriptional control of aquaporin-2 abundance by vasopressin in renal collecting duct principal cells. Am J Physiol Renal Physiol 2006; 290(1):F177.
9. Moeller HB, Olesen ET, Fenton RA. Regulation of the water channel aquaporin-2 by posttranslational modification. Am J Physiol Renal Physiol 2011; 300(5):F1062.
10. Rice WL, Zhang Y, Chen Y, et al. Differential, phosphorylation dependent trafficking of AQP2 in LLC-PK1 cells. PLoS One 2012; 7(2):e32843.
11. Eto K, Noda Y, Horikawa S, et al. Phosphorylation of aquaporin-2 regulates its water permeability. J Biol Chem 2010; 285(52):40777.
12. Stockand JD. Vasopressin regulation of renal sodium excretion. Kidney Int 2010; Nov;78(9):849.
13. Ecelbarger CA, Kim GH, Terris J, et al. Vasopressin-mediated regulation of epithelial sodium channel abundance in rat kidney. Am J Physiol Renal Physiol 2000; 279(1):F46.
14. Payne JA, Xu JC, Haas M, et al. Primary structure, functional expression, and chromosomal localization of the bumetanide-sensitive Na-K-Cl cotransporter in human colon. J Biol Chem 1995; 270: 17977.
15. Ares GR, Caceres PS, Ortiz PA. Molecular regulation of NKCC2 in the thick ascending limb. Am J Physiol Renal Physiol 2011; 301(6):F1143.
16. Kim GH, Ecelbarger CA, Mitchell C, et al. Vasopressin increases Na-K-2Cl cotransporter expression in thick ascending limb of Henle's loop. Am J Physiol 1999; 276(1 Pt 2):F96.
17. Blanchard A, Frank M, Wuerzner G, et al. Antinatriuretic effect of vasopressin in humans is amiloride sensitive, thus ENaC dependent. Clin J Am Soc Nephrol 2011; 6(4):753.
18. Sands JM, Blount MA, Klein JD. Regulation of renal urea transport by vasopressin. Trans Am Clin Climatol Assoc 2011; 122:82-92.
19. Koshimizu TA, Nasa Y, Tanoue A, et al. V1a vasopressin receptors maintain normal blood pressure by regulating circulating blood volume and baroreflex sensitivity. Proc Natl Acad Sci U S A 2006; 103(20):7807.
20. Aoyagi T, Izumi Y, Hiroyama M, et al. Vasopressin regulates the renin-angiotensin-aldosterone system via V1a receptors in macula densa cells. Am J Physiol Renal Physiol 2008; 295(1):F100.
21. Birumachi J, Hiroyama M, Fujiwara Y, et al. Impaired arginine-vasopressin-induced aldosterone release from adrenal gland cells in mice lacking the vasopressin V1A receptor. Eur J Pharmacol 2007; 566(1-3):226.
22. Ecelbarger CA, Nielsen S, Olson BR, et al. Role of renal aquaporins in escape from vasopressin-induced antidiuresis in rat. J Clin Invest 1997; 99(8):1852-63.
23. Murase T, Tian Y, Fang XY, Verbalis JG. Synergistic effects of nitric oxide and prostaglandins on renal escape from vasopressin-induced antidiuresis. Am J Physiol Regul Integr Comp Physiol 2003; 284(2):R354.
24. Kortenoeven ML, van den Brand M, Wetzels JF, Deen PM. Hypotonicity-induced reduction of aquaporin-2 transcription in mpkCCD cells is independent of the tonicity responsive element, vasopressin, and cAMP. J Biol Chem 2011; 286(15):13002.
25. Nagasaki H, Yokoi H, Arima H, et al. Overexpression of vasopressin in the rat transgenic for the metallothionein-vasopressin fusion gene. J Endocrinol 2002; 173(1):35.
26. Yokoi H, Nagasaki H, Tachikawa K, et al. Adaptation to sustained high plasma vasopressin in water and electrolyte homeostasis in the rat transgenic for the metallothionein-vasopressin fusion gene. J Endocrinol 2002; 173(1):23.
27. Tachikawa K, Yokoi H, Nagasaki H, et al. Altered cardiovascular regulation in arginine vasopressin-overexpressing transgenic rat. Am J Physiol Endocrinol Metab 2003; 285(6):E1161.
28. Hiyama TY, Matsuda S, Fujikawa A, et al. Autoimmunity to the sodium-level sensor in the brain causes essential hypernatremia. Neuron 2010; 66(4):508.